Comments
- No comments found

Deep learning is promising for solving a variety of issues in the pharmaceutical industry.
A few decades ago, the process of finding new drugs and developing them was confined to a group of medicinal chemists working in a lab. To get one medicine approved for use, there would be a significant investment of time and money in testing, validation, and synthesis methods. The development of numerous bioinformatics, pharmacy informatics, and cheminformatics tools has assisted in the acceleration of the drug discovery process due to advances in computational approaches mixed with an explosion in deep learning techniques.
Deep learning assists in the global initiative to curb the spread of infectious diseases and substantially speeds up drug discovery. Deep learning can quickly and accurately find drug candidates for novel, widespread diseases, improving the efficiency of screening antimicrobial compounds against a wide range of bacteria.
Drug discovery using deep learning allows for sophisticated image interpretation, molecular structure, function prediction, and the automated creation of novel chemical entities with specific features. Despite the increasing number of promising future applications, the underlying mathematical models are often challenging for the human mind to comprehend. To answer the requirement for a new narrative of the machine language of the molecular sciences, there is a desire for "explainable" deep learning approaches.
Software engineers and pharmacists have collaborated to build DL-based tools, predictive analytics and algorithms that may be utilized in the drug research and development procedure to harness the tremendous potential of deep learning models. Deep learning tools are classified based on their functionality as follows:
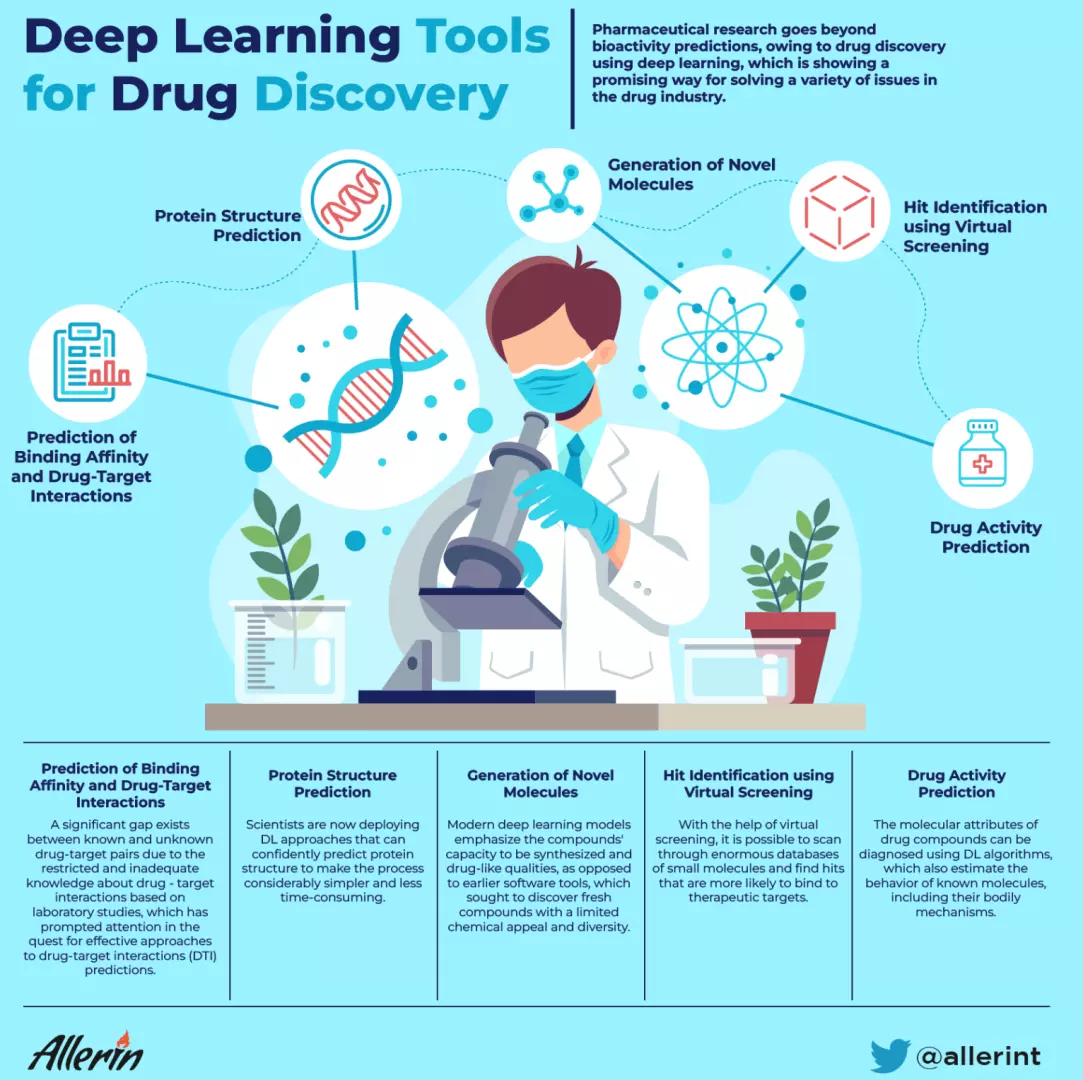
A significant gap exists between known and unknown drug-target pairs due to the restricted and inadequate knowledge about drug-target interactions based on laboratory studies, which has prompted attention in the quest for effective approaches to drug-target interactions (DTI) prediction.
Conventional means of DTI prediction have been hampered by financial and technological issues, but deep learning approaches are achieving the same results more efficiently.
Currently, ligand-based approaches, chemo-genomic approaches, text mining techniques, ML/ DL-based techniques and network-based strategies are the main computational methods employed in DTI prediction. Some examples of deep learning models in drug discovery for the prediction of DTI and binding affinity are DTI-CNN, DeepCPI, DeepDTA, Deep docking, etc.
Protein structure identification demands time and complex experimental techniques like X-ray crystallography, NMR spectroscopy, electron microscopy, etc. Competent computational methods are required to fill the vacuum left by many unidentified protein structures, which allows structural bioinformatics on a wide scale. Scientists are now deploying DL approaches that can confidently predict protein structure to make the process considerably simpler and less time-consuming. Alphafold and CASP are two of the most popular deep learning models used for this purpose.
Certain DL algorithms seek to identify novel drugs and drug-like substances. Modern deep learning models emphasize the compounds' capacity to be synthesized and drug-like qualities, as opposed to earlier software tools, which sought to discover fresh compounds with a limited chemical appeal and diversity.
With the help of virtual screening, it is possible to scan through enormous databases of small molecules and find hits that are more likely to bind to therapeutic targets. Distinct characteristics of a chemical substance and the target are used in the virtual screening technique. Numerous studies have demonstrated that DL algorithms, such as Similarity Search and DeepVS, perform substantially better in virtual screening than other ML algorithms, particularly when used in the de novo molecular design process, which uses sequential data to create molecules with the required characteristics.
The molecular attributes of drug compounds can be diagnosed using DL algorithms, which also estimate the behavior of known molecules, including their bodily mechanisms. The cost and time required will be greatly reduced by applying the semi-supervised learning technique to anticipate the therapeutic application of pharmaceuticals from their structural formulas. Due to its logical data adjustment and small number of labeled data, Multicon is a DL-based tool with a higher rate of drug activity estimation than other semi-supervised learning algorithms.
A long and costly process, drug discovery has always faced challenges in moving a drug prospect into clinical testing. Deep learning has significantly enhanced the operation's success rate, which minimizes the overall research cost. With the massive advancements in the DL toolset, it can yield considerable benefits. Modern DL techniques will certainly be highly valued in the upcoming era of big data search and analysis for drug design and discovery after considering the recent success of such methodology and its use by pharmaceutical corporations.
Naveen is the Founder and CEO of Allerin, a software solutions provider that delivers innovative and agile solutions that enable to automate, inspire and impress. He is a seasoned professional with more than 20 years of experience, with extensive experience in customizing open source products for cost optimizations of large scale IT deployment. He is currently working on Internet of Things solutions with Big Data Analytics. Naveen completed his programming qualifications in various Indian institutes.
BBN Times connects decision makers to you. Experts in their fields, worth listening to, are the ones who write our articles. We believe these are the real commentators of the future. We quickly and accurately deliver serious information around the world. BBN Times provides its readers human expertise to find trusted answers by providing a platform and a voice to anyone willing to know more about the latest trends. Stay tuned, the revolution has begun.
Leave your comments
Post comment as a guest